






Source: Science Daily news
by: Georgia Institute of Technology. Researchers are using machine learning to ultimately find ways to build more capable capacitors.
Capacitors, given their high energy output and recharging speed, could play a major role in powering the machines of the future, from electric cars to cell phones. But the biggest hurdle for these energy storage devices is that they store much less energy than a battery of similar size.
Researchers at Georgia Institute of Technology are tackling that problem in a novel way, using machine learning to ultimately find ways to build more capable capacitors. The method, which was described in February 18 in the journal npj Computational Materials and sponsored by the U.S. Office of Naval Research, involves teaching a computer to analyze at an atomic level two materials that make up some capacitors: aluminum and polyethylene.
The researchers focused on finding a way to more quickly analyze the electronic structure of those materials, looking for features that could affect performance.
“The electronics industry wants to know the electronic properties and structure of all of the materials they use to produce devices, including capacitors,” said Rampi Ramprasad, a professor in the School of Materials Science and Engineering.
Take a material like polyethylene: it is a very good insulator with a large band gap — an energy range forbidden to electrical charge carriers. But if it has a defect, unwanted charge carriers are allowed into the band gap, reducing efficiency, he said.
“In order to understand where the defects are and what role they play, we need to compute the entire atomic structure, something that so far has been extremely difficult,” said Ramprasad, who holds the Michael E. Tennenbaum Family Chair and is the Georgia Research Alliance Eminent Scholar in Energy Sustainability. “The current method of analyzing those materials using quantum mechanics is so slow that it limits how much analysis can be performed at any given time.”
Ramprasad and his colleagues, who specialize in using machine learning to help develop new materials, used a sample of data created from a quantum mechanics analysis of aluminum and polyethylene as an input to teach a powerful computer how to simulate that analysis.
Analyzing the electronic structure of a material with quantum mechanics involves solving the Kohn-Sham equation of density functional theory, which generates data on wave functions and energy levels. That data is then used to compute the total potential energy of the system and atomic forces.
Using the new machine learning method produces similar results eight orders of magnitude faster than using the conventional technique based on quantum mechanics.
“This unprecedented speedup in computational capability will allow us to design electronic materials that are superior to what is currently out there,” Ramprasad said. “Basically we can say, ‘Here are defects with this material that will really diminish the efficiency of its electronic structure.’ And once we can address such aspects efficiently, we can better design electronic devices.”

left image: Rampi Ramprasad, a professor in the School of Materials Science and Engineering, holds an aluminum-based capacitor. Credit: Allison Barnes, Georgia Tech
While the study focused on aluminum and polyethylene, machine learning could be used to analyze the electronic structure of a wide range materials. Beyond analyzing electronic structure, other aspects of material structure now analyzed by quantum mechanics could also be hastened by the machine learning approach, Ramprasad said.
“In part we selected aluminum and polyethylene because they are components of a capacitor, but it also allowed us to demonstrate that you can use this method for vastly different materials, such as metals that are conductors and polymers that are insulators,” Ramprasad said.
The faster processing allowed by the machine learning method would also enable researchers to more quickly simulate how modifications to a material will impact its electronic structure, potentially revealing new ways to improve its efficiency.
featured image:
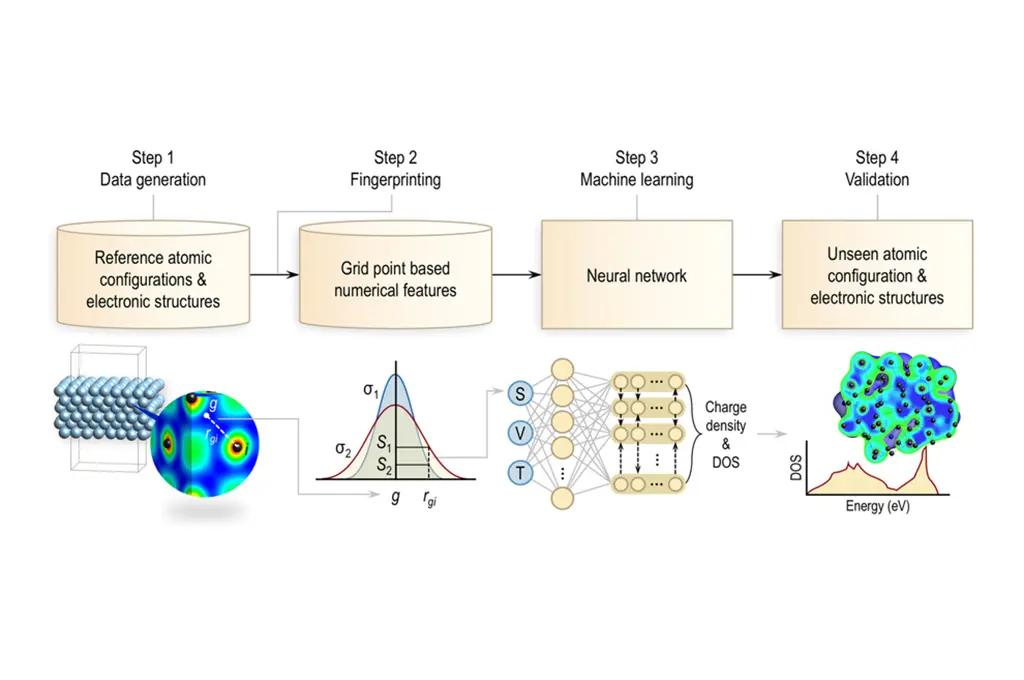
Overview of the process used to generate surrogate models for the charge density and density of states. The first step entails the generation of the the training dataset by sampling random snapshots of molecular dynamics trajectories. First-principles calculations were then performed on these systems (shown in Figure S1) to obtain the training atomic configurations, charge densities, and local density of states. The scalar (S), vector (V), and tensor (T) fingerprint invariants are mapped to the local electronic structure at every grid-point. For the charge density, this mapping is achieved using a simple fully connected neural network with one output neuron. The LDOS spectrum, on the other hand, is learned via a recurrent neural network architecture, wherein the LDOS at every energy window is represented as a single output neuron (linked via a recurrent layer to other neighboring energy windows). The trained model is then used to predict the electronic structure (i.e, DOS and charge density) of an unseen configuration.
featured image source: Georgia Institute of Technology
Story Source:
Science Daily article. Materials provided by Georgia Institute of Technology. Note: Content may be edited for style and length.
Journal Reference:
Anand Chandrasekaran, Deepak Kamal, Rohit Batra, Chiho Kim, Lihua Chen, Rampi Ramprasad. Solving the electronic structure problem with machine learning. npj Computational Materials, 2019; 5 (1) DOI: 10.1038/s41524-019-0162-7