



Researchers from Nanjing University in China published an article about upgrade of EDLC supercapacitors by a lignin-derived hierarchical porous carbon and a Li⁺ weakly solvating electrolyte to achieve 4.0 V operation with high energy density and strongly suppressed self-discharge.
Introduction
Electrochemical double-layer supercapacitors (EDLCs) combine high power density and long cycle life but remain constrained by low energy density and pronounced self-discharge, which limits their use in energy-intensive applications such as electric vehicles and grid storage. Because the stored energy scales with the square of the cell voltage, extending the operating window from the commercial standard of about 2.7 V to around 4.0 V is an attractive route to boost energy density, but conventional aqueous and organic electrolytes tend to decompose at such elevated voltages.
Previous strategies to reach higher voltages, including ionic liquid electrolytes and high-concentration electrolytes, improve electrochemical stability but suffer from high viscosity, sluggish ion transport, higher cost, and aggravated parasitic reactions that accelerate self-discharge. In parallel, lignin-derived porous carbons have emerged as sustainable EDLC electrodes, but effective high-voltage operation requires precise matching between pore structure, ion solvation, and electrolyte chemistry. This article addresses these coupled limitations through a co-design of a lignin-derived hierarchical porous carbon (HPC) electrode and a Li⁺-based weakly solvating electrolyte formulated with sulfolane (SUL) and a fluorinated ether diluent.
Key points
- Demonstration of a lignin-derived hierarchical porous carbon (HPC) with high surface area (≈2844 m² g⁻¹) and dominant sub-nanometer pores (~0.8 nm), tailored for solvated Li⁺ and BF₄⁻ ions.
- Formulation of a LiBF₄-based weakly solvating electrolyte using sulfolane (SUL) and a non-coordinating fluorinated ether diluent (TTE) to combine high oxidative stability with reduced viscosity and fast ion transport.
- Achieved specific capacitance of 139 F g⁻¹ at 4.0 V in symmetric HPC-based EDLCs with excellent rate performance (92 F g⁻¹ at 10 A g⁻¹).
- Realized a maximum energy density of 77.4 Wh kg⁻¹, exceeding typical carbon-based symmetric supercapacitors (≈25–35 Wh kg⁻¹), while operating stably at 4.0 V.
- Demonstrated robust cycling stability with over 90% capacitance retention after 10,000 charge–discharge cycles at 10 A g⁻¹ and near-unity Coulombic efficiency.
- Quantitatively analyzed self-discharge via open-circuit voltage decay and leakage current, showing that the weakly solvating electrolyte markedly suppresses both potential-driven ion redistribution and activation-controlled parasitic reactions.
- XPS and LSV measurements confirmed reduced electrolyte decomposition and the formation of a more stable electrode–electrolyte interface in the weakly solvating electrolyte compared to conventional TEABF₄/PC systems.
- Validated that geometric matching between sub-nanometer pores and solvated Li⁺ dimensions is essential for maximizing charge storage, outperforming both under-activated and over-activated carbons as well as commercial YP-50F.
Extended summary
The work targets two fundamental bottlenecks of EDLCs: low energy density and severe self-discharge at elevated voltages. Conventional aqueous electrolytes are limited to about 1.2 V by water decomposition, while widely used organic electrolytes based on TEABF₄ in acetonitrile or propylene carbonate start to degrade beyond about 2.7 V, leading to electrolyte breakdown and rapid self-discharge. Approaches relying on ionic liquids or high-concentration electrolytes can extend the voltage window but introduce excessive viscosity, increased cost, and more intense parasitic reactions at the carbon surface. In parallel, literature has shown that the capacitance of EDLCs is highly sensitive to the size compatibility between solvated ions and the carbon pore network, especially for nanoporous carbons. Building on this, the authors propose a synergistic strategy combining a pore-size-engineered lignin-derived carbon with a weakly solvating Li⁺ electrolyte to simultaneously improve voltage stability, energy density, and self-discharge behavior.
The electrode material is obtained from industrial lignin using a KOH activation–pyrolysis route in a tube furnace under nitrogen. Lignin is mixed with KOH at a mass ratio of 1:2, heated first to 250 °C, then to 800 °C, and subsequently washed with water and HCl to remove inorganic residues, yielding a highly porous carbon denoted HPC. XRD shows broad (002) and (110) reflections characteristic of disordered carbon, while Raman spectroscopy exhibits an ID/IG ratio of 0.97, indicating a highly defective, non-graphitic structure. Nitrogen adsorption–desorption measurements give a very high specific surface area of 2844 m² g⁻¹ and a combined Type I/IV isotherm, implying abundant micropores and mesopores. Pore size analysis reveals a dominant peak around 0.8 nm, which is close to the solvated size of Li⁺ in sulfolane (~0.8 nm) and BF₄⁻ (~0.6 nm), enabling efficient ion confinement. Scanning electron microscopy reveals a three-dimensional, interconnected porous network that contrasts with the dense morphology of commercial YP-50F carbon, and EDS/XPS confirm uniformly distributed carbon and oxygen with surface C–O and C=O functionalities that enhance wettability.
To probe the role of activation, two additional carbons are prepared with KOH:lignin ratios of 1:1 (HPCinsu) and 3:1 (HPCexce). HPCinsu exhibits smaller micropores (~0.6 nm) that are too tight for solvated Li⁺, while HPCexce shows a broad 1–8 nm pore distribution with reduced sub-nanometer fraction. When tested in Li⁺-based electrolytes, HPC delivers a specific capacitance of 133 F g⁻¹ at 2.7 V, outperforming HPCinsu and HPCexce as well as commercial YP-50F (≈90 F g⁻¹), which is attributed to the optimal pore–ion size matching. In contrast, TEA⁺ cations with a solvated size of 1.3–1.4 nm show poor utilization of the 0.8 nm pores, resulting in shorter discharge times and lower capacitance, highlighting the importance of ion–pore compatibility.
On the electrolyte side, the authors first investigate LiBF₄ in sulfolane at varying concentrations to extend the voltage window. Increasing the LiBF₄ concentration enhances electrochemical stability via a high-concentration electrolyte effect, but at the cost of higher viscosity and reduced capacitance. Cyclic voltammetry and galvanostatic charge–discharge reveal that 4 M LiBF₄/SUL gives the best trade-off, with a specific capacitance of 97.5 F g⁻¹ and Coulombic efficiency of 94%; higher concentrations further reduce capacitance, whereas lower concentrations exacerbate electrolyte decomposition and lower Coulombic efficiency. To circumvent the viscosity issue, the electrolyte is transformed into a weakly solvating system by adding 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE) as a non-coordinating diluent. TTE is miscible with sulfolane but does not bind Li⁺, thereby preserving local Li⁺–SUL clusters while diluting the bulk and lowering viscosity.
The optimized weakly solvating electrolyte consists of 4 M LiBF₄ in SUL mixed with TTE at SUL:TTE = 1:2 by volume. In this medium, the HPC electrodes exhibit nearly rectangular CV curves up to 4.0 V, longer discharge times, and lower charge-transfer resistance, indicating more ideal capacitive behavior and faster ion kinetics. Galvanostatic data show a maximum specific capacitance of 139 F g⁻¹, with 92 F g⁻¹ retained even at 10 A g⁻¹, confirming excellent rate capability. Long-term cycling at 10 A g⁻¹ over 10,000 cycles reveals capacitance retention above 90% and almost unity Coulombic efficiency, demonstrating that the co-designed electrode–electrolyte system is stable under high-voltage, high-rate conditions. The Ragone plot places this device at an energy density of 77.4 Wh kg⁻¹, more than double that of typical symmetric carbon supercapacitors and among the best reported carbon-based systems.
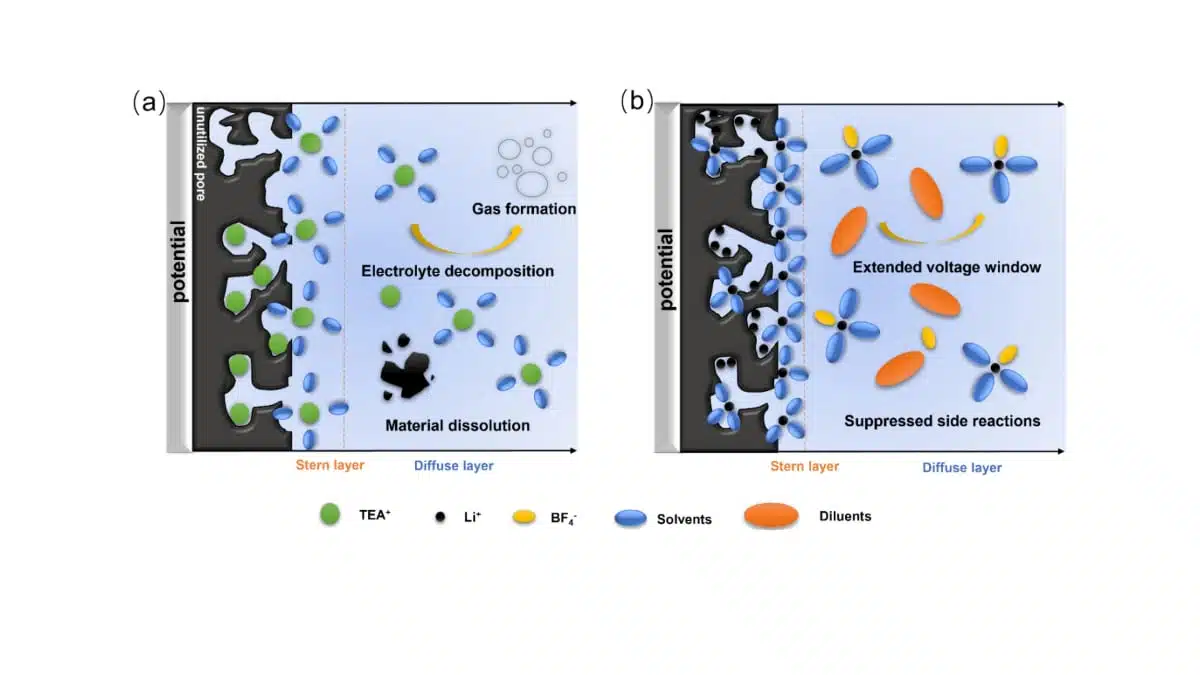
Self-discharge behavior is analyzed in detail because it is a critical limiting factor for practical deployment. The authors distinguish two main regimes: an initial fast voltage drop driven by potential-driven ion redistribution within the porous network, and a slower long-term decay governed by parasitic Faradaic reactions. Open-circuit voltage decay measurements after charging to 4.0 V show that the weakly solvating electrolyte exhibits the slowest initial voltage drop, suggesting that it forms a more ordered Helmholtz layer that resists ion redistribution. To quantify the contributions of different processes, the OCV curves are fitted with a model that decomposes the decay into an initial Ohmic drop, diffusion-controlled Faradaic reactions (DCFR), and activation-controlled Faradaic reactions (ACFR). The fitted parameters indicate a markedly reduced ACFR component in the weakly solvating electrolyte compared to both 1 M LiBF₄/SUL and 4 M LiBF₄/SUL, pointing to effectively suppressed slow parasitic reactions at high voltage.
Leakage current measurements provide complementary evidence: cells with the weakly solvating electrolyte show the lowest steady-state leakage current at both 2.7 and 4.0 V, confirming reduced continuous charge loss. XPS analysis of HPC electrodes after 10,000 cycles in different electrolytes further elucidates the interfacial chemistry. Electrodes cycled in conventional 1 M TEABF₄/PC display a strong C–F signal and increased C=O content in the C 1s spectrum, indicative of substantial electrolyte decomposition and the formation of fluorinated surface species. In contrast, electrodes cycled in 4 M LiBF₄/SUL:TTE retain their original C–O and C=O features without detectable C–F peaks, demonstrating that the weakly solvating electrolyte preserves the carbon surface and limits parasitic reactions. Linear sweep voltammetry corroborates these findings, showing that TEABF₄/PC begins to oxidize above about 2.5 V, while the TTE-containing electrolyte remains stable up to around 4.2 V.
Overall, the study shows that combining a lignin-derived HPC with sub-nanometer pores and a carefully formulated weakly solvating Li⁺ electrolyte yields a device with high capacitance, extended 4.0 V operation, high energy density, and strongly suppressed self-discharge. The key mechanisms are geometric matching between pore size and solvated ion size, which maximizes double-layer charge storage, and a weakly solvating, fluorinated diluent environment that reduces viscosity, accelerates ion transport, and stabilizes the electrode–electrolyte interface against oxidative degradation.
Conclusion
This work demonstrates a sustainable, high-performance EDLC concept by co-designing a lignin-derived hierarchical porous carbon and a LiBF₄/SUL–TTE weakly solvating electrolyte to overcome the intrinsic voltage and self-discharge limitations of conventional supercapacitors. The resulting symmetric devices combine high specific capacitance, an unprecedented 4.0 V operating window, energy density of 77.4 Wh kg⁻¹, and excellent cycling stability with strongly reduced self-discharge and leakage.
The main limitations include the need for controlled synthesis of the HPC to finely tune pore size distribution and the use of a relatively specialized fluorinated ether diluent whose large-scale cost and environmental footprint will require further assessment. Future work could explore scaling the lignin-based carbon synthesis, optimizing electrolyte composition for other cations or solvents, integrating this design into larger-format devices, and extending the weakly solvating electrolyte strategy to hybrid or pseudocapacitive systems.
References
- Shichao Zhang, Shenglin Liu, Suyang Si, Keqi Zeng, Chenxin Cai, Xiangzhou Yuan, Yawen Tang, Feng Gong, Hualin Ye, “Lignin-derived hierarchical porous carbons enabling high-voltage electrochemical capacitors with low self-discharge,” Carbon Research, Volume 5, Article 11, 2026.